PENEPMA (Secondary Fluorescence Profile) Calculations
From EPMA Xtreme Probe
PENEPMA (Secondary Fluorescence Profile) Calculations
This menu provides an easy graphical user interface to calculate secondary fluorescence profiles using Penepma12 FORTRAN executables.
This menu is available from both CalcZAF program and Probe for EPMA. Simply select the Standards | Standard Database menu to launch the Standard application. Accept the default (or other) standard database. From the Standard application click the Analytical | PENEPMA (Secondary Fluorescence Profile) Calculations menu.
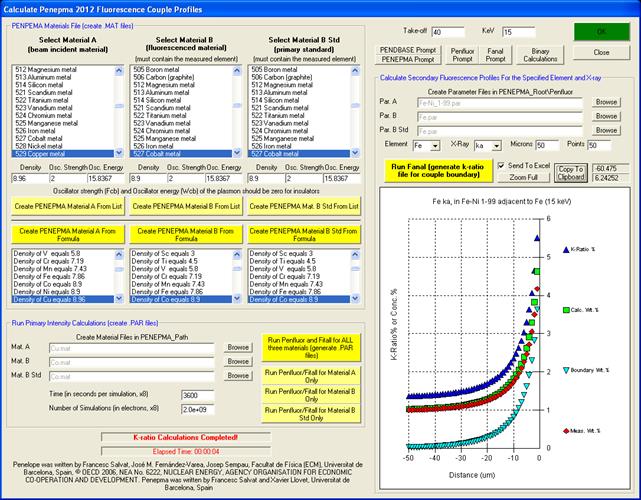
The general procedure is to select two materials, one each for the two phases of the beam incident and boundary materials and a third phase, usually a pure element as a standard for the specified element and x-ray, for the calculation of k-ratio intensities. If the standard composition selected is NOT a pure element then the k-ratio intensities will be “raw” k-ratios as opposed to elemental k-ratios.
Note that material A is always on the left side of the geometry file, material B is on the right side of the geometry file and the electron beam is always incident on material A. Note that this is the opposite orientation of the normal couple.geo file used by Penepma for Monte Carlo calculations.
The element being fluoresced may only be present in material B or it may be in both material A and material B (or in neither material though this will not be as interesting).
If the same (compound) material is specified for both material A and material B then the calculation merely calculates the normal sample fluorescence (from both characteristic and continuum fluorescence) and also the full bulk matrix correction.
After calculating the three material files (or selecting them using the Browse buttons), click the Run Penfluor and Fitall buttons to calculate the .par files for use by Fanal.exe. The default time for each .par file is 8 hours (3600 seconds for each electron energy for 8 electron energies). The default lowest electron energy is 1 keV and the highest electron energy is 50 keV. The default is to calculate photon energies down to 1 keV although this default may be modified by editing the PenepmaMinimumElectronEnergy keyword in the [software] section of the PROBEWIN.INI file to obtain intensities for lower energy x-ray lines.
Once the PAR files are calculated one may use the Run Fanal button to run program Fanal.exe to calculate k-ratios for the boundary or matrix specified for Par. A, Par. B and Par. B Std. The following will describe the basic functions in more detail.
![]() PENEPMA Material Files (create .MAT files)
PENEPMA Material Files (create .MAT files)
![]() Run Primary Intensity Calculations (create .PAR files)
Run Primary Intensity Calculations (create .PAR files)
![]() Calculate Secondary Fluorescence Profiles
Calculate Secondary Fluorescence Profiles
![]() Assumption Of Bulk Matrix Corrections
Assumption Of Bulk Matrix Corrections
![]() Compositions Exhibiting Self Fluorescence Adjacent to a
Non-Fluorescing Phase
Compositions Exhibiting Self Fluorescence Adjacent to a
Non-Fluorescing Phase