APF (Area Peak Factor)
The APF values are used to correct light element intensities (e.g., O ka, N ka, C ka and B ka) for peak shape (and shift) effects that can affect quantitative analysis. Since the degree of shape and shift is to a first order dependent only on the element which is bonded to the light element (and to a lesser extent on the coordination or valence of the element) the binary effect (as measured on pure oxide, carbide, nitride or boride, etc. compounds) is first characterized using the following equation.
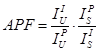
where:
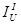 is the
integrated intensity for the “unknown”
is the
integrated intensity for the “unknown”
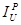 is the peak
intensity for the “unknown”
is the peak
intensity for the “unknown”
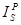 is the peak
intensity for the “standard”
is the peak
intensity for the “standard”
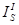 is the
integrated intensity for the “standard”
is the
integrated intensity for the “standard”
The total peak shape/shift effect can be summed together to obtain an “average” APF for a complex compound by summing the weight fractions of the “binary” APF values.
The “binary” APF values are taken from the EMPAPF.DAT file and the default values were determined by Bastin on a Cameca microprobe. Since the values depend on the spectrometer and crystal resolution, each analyst should measure the values on their own instrument by performing careful wavelength scans on both “standard” reference peaks and “unknown” reference peaks. For example, if it necessary to correct for oxygen peak shape changes in an unknown compound containing both Si and Al bonded to oxygen (SiO2 and Al2O3), the user should measure a reference oxygen peak, usually Fe2O3 (used by Bastin) or MgO (used by Donovan) and also the oxygen peaks on SiO2 and Al2O3.
In the example given, MgO or Fe2O3 are the standard
reference 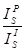 and Al2O3 and SiO2 are the
binary references
and Al2O3 and SiO2 are the
binary references 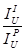 .
.
The Model Backgrounds dialog provides a quick way to determine integrated peak intensities and provides the P/I and I/P values which can also be output to a text file, though more sophisticated applications will be necessary if other peaks are present (e.g., Igor).