ZAF Matrix Corrections
From EPMA Probe
ZAF Matrix Corrections
ZAF matrix corrections (included here are also the newer Phi-Rho-Z corrections) are a fundamental calculation of the x-ray intensities based on physical models of the atomic and electron interaction.
It is the most accurate way available to correct for matrix effects without using an empirical calibration curve. It is also very calculation intensive, but for modern CPU's this is a non-issue, except possibly when analyzing extremely large numbers of elements or samples. The following expression summarizes the ZAF correction :
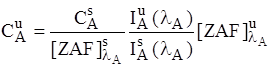
where : 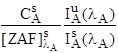 is the
unknown k-ratio and
is the
unknown k-ratio and
![]() is the ZAF correction factor of the unknown
is the ZAF correction factor of the unknown
and
: ![]() is the unknown intensity for element A at λ
is the unknown intensity for element A at λ
![]() is the standard intensity for element A at λ
is the standard intensity for element A at λ
![]() is the concentration of the element in the standard
is the concentration of the element in the standard
![]() is the ZAF correction for the element in the standard
is the ZAF correction for the element in the standard
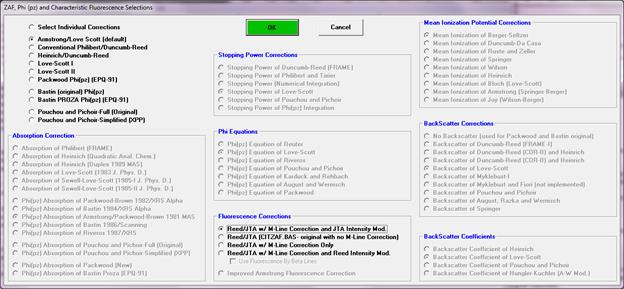
Probe for EPMA includes the most commonly used corrections schemes for ZAF, Phi-Rho-Z and PAP matrix corrections. All these options are available with a single mouse click allowing the user to switch quickly between correction schemes in order to compare the results obtained. The main choices are:
1 … Select Individual Corrections
2 … Armstrong/Love Scott (default)
3 … Conventional Philibert/Duncumb-Reed
4 … Heinrich/Duncumb-Reed
5 … Love-Scott I
6 … Love-Scott II
7 … Packwood Phi(pz) (EPQ-91)
8 … Bastin (original) Phi(pz)
9 … Bastin PROZA Phi(pz) (EPQ-91)
10 … PAP- Pouchou and Pichoir – Full (Original)
11 … PAP- Pouchou and Pichoir – Simplified (XPP)
The default selection is the Armstrong/Love-Scott Phi-Rho-Z option. Note that depending on the actual mass absorption coefficients, atomic weights, x-ray emission and absorption edge energies and fluorescent yields used, the analytical results can vary slightly. The files used for these purposes are documented elsewhere (see CalcZAF.Exe) but are noted here as well:
XLINE.DAT Default x-ray line energies (copy XLINE1.DAT or XLINE2.DAT to this name)
XEDGE.DAT Default x-ray edge energies (copy XEDGE1.DAT or XEDGE2.DAT to this name)
XFLUR.DAT Default fluorescent yields (copy XFLUR1.DAT or XFLUR2.DAT to this name)
The following are the original files from two different sources that should be renamed to be utilized by the Probe for EPMA applications:
XLINE1.DAT Johnson and White x-ray line energies (no beta energies)
XEDGE1.DAT Johnson and White x-ray edge energies (no beta energies)
XFLUR1.DAT Armstrong ELEMINFO.DAT Fluorescent yields (no beta yields)
XLINE2.DAT Armstrong ELEMINFO.DAT x-ray line energies
XEDGE2.DAT Armstrong ELEMINFO.DAT x-ray edge energies
XFLUR2.DAT Armstrong ELEMINFO.DAT fluorescent yields (no beta yields)
The "Select Individual Corrections" option may be used to specifically select individual correction procedures from a large array of options. Note that many correction procedures will only work with other specific choices. The following is taken from John Armstrong's CITZAF documentation on how to select among the various matrix correction options :